Given a reasonably equilibrated configuration we can try enhanced sampling of the Volmer step. To generate a simple plumed.dat file, you can use appa plumed volmer. View the structure and note the (zero-based) indices of the hydrogen atom that you want to transfer, the nearest surface atom, and the most accessible oxygen atom. Then use the command as follows:
Usage: appa plumed volmer [OPTIONS]
Write plumed.dat input file for a Volmer step calculation.
Options:
-o, --oxygen_id INTEGER
-h, --hydrogen_id INTEGER
-s, --surface_id INTEGER
--stride INTEGER How often to print to COLVAR [default: 10]
--help Show this message and exit.For example:
appa plumed volmer -o 107 -s 56 -h 64You can then setup a molecular dynamics run with appa lammps, such as
appa lammps initial.xyz --architecture grace --model ~/plumed-test/train/seed/1/final_model --steps 800000 --plumed-file plumed.datYou can also write different PLUMED files, such as umbrella sampling of a reaction coordinate suggested by Kronberg & Laasonen, and Santos et al..
With some Python code you can define the initial :
pos = atoms.positions
d_OH = np.linalg.norm(pos[oxygen_id, :] - pos[hydrogen_id, :])
d_MH = np.linalg.norm(pos[surface_id, :] - pos[hydrogen_id, :])
xi0 = d_OH - d_MHUpon defining the number of warmup timesteps, a kappa and a cv_target, you can use Python string formatting to write part of a PLUMED file such as
# Reaction coordinate xi = d(O-H) - d(Pt-H)
xi: COMBINE ARG=d_OH,d_MH COEFFICIENTS=1,-1 PERIODIC=NO
# Harmonic umbrella restraint
restraint: MOVINGRESTRAINT ...
ARG=xi
STEP0=0 AT0={xi0:.2f} KAPPA0=0.0
STEP1={warmup} AT1={cv_target:.2f} KAPPA1={kappa}
...However, in my experience this results in the proton diffusing along the surface, creating a large and , resulting in a but a structure very different from the expected transition state. Adding more training data around the TS might help.
Analysis¶
To get a free-energy surface from metadynamics, you can use
module load 2025
module load PLUMED/2.9.4-foss-2025a
plumed sum_hills --hills path/to/my/HILLS
module purge(since I’m not sure how to call the plumed that was built upon LAMMPS install).
You can plot it to get a result like this:
import numpy as np
import matplotlib.pyplot as plt
plt.style.use('data/lucas.mplstyle')
data = np.loadtxt('data/fes.dat', comments='#')
cv1 = data[:,0]
cv2 = data[:,1]
energy = data[:,2]
plt.figure(figsize=(4,3))
plt.tricontourf(cv1, cv2, energy, levels=10, cmap='magma')
plt.colorbar()
plt.xlabel('d_OH')
plt.ylabel('d_MH')
plt.show()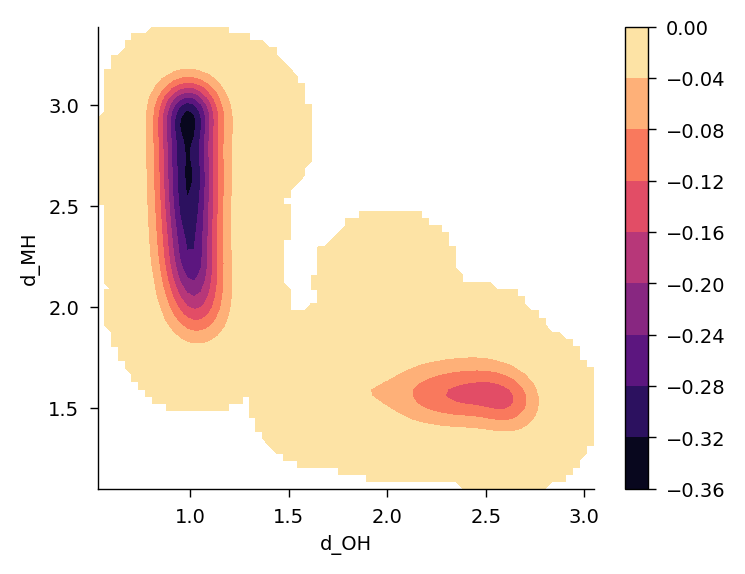
Probably limited training data, limited metadynamics sampling time, and limited training time all contributes to this result not being amazing. The proton crossed the energy barrier once in this run. But it still gives an idea of what is possible with MLIPs.
- Kronberg, R., & Laasonen, K. (2021). Reconciling the Experimental and Computational Hydrogen Evolution Activities of Pt(111) through DFT-Based Constrained MD Simulations. ACS Catalysis, 11(13), 8062–8078. 10.1021/acscatal.1c00538
- Santos, E., Aradi, B., van der Heide, T., & Schmickler, W. (2024). Free energy curves for the Volmer reaction obtained from molecular dynamics simulation based on quantum chemistry. Journal of Electroanalytical Chemistry, 954, 118044. 10.1016/j.jelechem.2024.118044